Majalah Farmasetika Jurnal Ilmiah Nasional Terakreditasi SINTA 3
Majalah Farmasetika Jurnal Ilmiah Nasional Terakreditasi SINTA 3
Artikel Terkait


Majalah Farmasetika, 10 (6) 2025, 485-505
https://doi.org/10.24198/mfarmasetika.v10i6.67440
Artikel Penelitian
LAria Aristokrat1*, Anis Yohana Chaerunisaa2
1Program Studi Profesi Apoteker, Fakultas Farmasi, Universitas Padjadjaran, Jatinangor, Jawa Barat, Indonesia 45363
2Departemen Farmasetika dan Teknologi Farmasi, Fakultas Farmasi, Universitas Padjadjaran, Jatinangor, Jawa Barat, Indonesia 45363
*E-mail : aria21001@mail.unpad.ac.id
(Submit 17/10/2025, Revisi 22/10/2025, Diterima 14/11/2025, Terbit 04/12/2025)
Abstrak
Kontaminasi silang pada sarana produksi adalah hal yang sangat dihindari sehingga perlu dilakukannya pembersihan yang sudah divalidasi karena residu yang tersisa dapat menjadi cemaran. Namun, terdapat risiko yang dapat terjadi selama dilakukannya validasi pembersihan. Oleh karena itu penting untuk mengkaji risiko tersebut. Penelitian ini akan mengkaji resiko yang dapat terjadi pada setiap tahapan validasi pembersihan dengan metode Failure Mode and Effects Analysis (FMEA). Penelitian dilakukan dengan cara observasional dan wawancara langsung di salah satu industri farmasi di Kota Bandung. Tahapan kajian risiko dimulai dengan mengidentifikasi risiko, menganalisis risiko berdasarkan tingkat keparahan, memberikan skor sehingga didapatkan nilai RPN, serta mengevaluasi risiko. Dari hasil kajian risiko, didapatkan sebanyak 26 risiko dengan 16 risiko minor, 4 risiko moderat dan 6 risiko mayor. Risiko yang termasuk dalam kategori mayor dibuat tindakan pengendalian risiko untuk mengurangi risiko terjadinya kegagalan dalam validasi pembersihan. Industri farmasi dapat melakukan tindakan pencegahan dan pengendalian dari hasil kajian risiko ini seperti pengecekan terhadap sarana penunjang kritis yang ada, penentuan marker, pelaksanaan proses validasi, serta mempertimbangkan untuk pembersihan secara otomatis.
Kata kunci: Evaluasi risiko, industri farmasi, kontaminasi silang
Teks Lengkap:
Pendahuluan
Kontaminasi silang merupakan proses ketika suatu bahan, produk, atau mikroorganisme yang tidak diinginkan secara tidak sengaja berpindah dari satu tempat ke tempat lain, sehingga dapat mencemari bahan atau produk (1). Dalam industri farmasi, kontaminasi silang menjadi perhatian utama karena dapat berdampak buruk pada kualitas, keamanan, dan efikasi obat, yang pada akhirnya dapat membahayakan pasien. Apabila pembersihan tidak dilakukan secara tepat maka akan ada kemungkinan residu yang dapat mencemari produk selanjutnya (2). Salah satu cara untuk mencegah kontaminasi silang adalah dengan melakukan proses pembersihan yang tepat.
Pembersihan pada area produksi merupakan salah satu hal yang penting dan kritis dalam industri farmasi. Terdapat dua macam pembersihan yaitu Clean-in-Place (CIP) dan Clean-out-of-Place (COP). CIP adalah proses otomatis yang membersihkan peralatan besar seperti tangki dan pipa tanpa perlu dibongkar. Kelebihan dari CIP adalah konsistensi tinggi, efisiensi waktu, dan pengurangan risiko kontaminasi manusia. Namun, terdapat kelemahan dari CIP yaitu membutuhkan biaya yang tinggi dan inspeksi visual yang terbatas. Sebaliknya, COP melibatkan pembongkaran komponen untuk dibersihkan secara manual atau semi-otomatis di area terpisah, memungkinkan inspeksi visual yang mendalam dan fleksibilitas untuk bagian yang sulit dijangkau, tetapi kekurangannya adalah intensitas tenaga kerja yang tinggi, downtime yang lebih lama, dan potensi inkonsistensi serta risiko kontaminasi ulang dari penanganan manual (3,4).
Untuk memastikan prosedur pembersihan yang dilakukan sudah tepat maka perlu dilakukannya validasi pembersihan. Validasi pembersihan adalah proses terdokumentasi yang memberikan bukti objektif bahwa prosedur pembersihan yang dilakukan untuk proses produksi secara konsisten dan efektif menghilangkan residu produk sebelumnya, agen pembersih, dan mikroorganisme hingga memenuhi kriteria seperti batas jumlah mikroba maupun koloni (5).
Validasi pembersihan dalam industri farmasi tidak luput dari risiko yang dapat terjadi terlebih dalam pembersihan yang dilakukan secara manual oleh operator tanpa menggunakan instrumen khusus yang terdapat pada alat. Contoh risiko yang dapat terjadi adalah kemungkinan pembersihan yang dilakukan oleh operator berbeda sehingga hasil pembersihan menjadi berbeda dan tidak valid. Hal ini akan mengakibatkan masih terdapatnya residu yang berdampak terjadinya kontaminasi terhadap produk selanjutnya. Oleh karena itu, kajian risiko diperlukan untuk menganalisis risiko yang dapat terjadi serta mengurangi risiko tersebut agar tidak berakibat fatal. Hal ini diperlukan untuk membantu dalam membuat keputusan yang lebih tepat dan efektif dengan memahami potensi risiko dan dampaknya (6,7). Kajian risiko berpedoman pada ICH Q9 dan CPOB tahun 2024 Aneks 13 (8,9) yang dapat digunakan untuk audit internal sebagai three line defense yang paling awal. Metode yang umum digunakan untuk mengidentifikasi risiko adalah dengan menggunakan 5 Why dan Ishikawa Fishbone, sedangkan untuk menilai risiko dengan menggunakan Failure Mode and Effects Analysis (FMEA), Failure Mode Effect and Criticality Analysis (FMECA), dan Hazard Analysis and Critical Control Points (HACCP) (10,11).
Oleh karena itu, penting untuk melakukan kajian risiko dalam validasi pembersihan terkhusus dengan pembersihan secara manual. Mengingat validasi pembersihan akan memberikan pengaruh terhadap proses pembersihan yang menjadi salah satu parameter penting di dalam industri farmasi. Diharapkan kajian ini dapat menjadi pedoman dan pengambilan keputusan bagi industri farmasi untuk meminimalisasi risiko yang dapat terjadi.
Metode
Penilaian risiko dilakukan secara observasional dan wawancara dengan personel yang terlibat selama proses validasi pembersihan di salah satu industri farmasi di Kota Bandung. Hasil penelitian berupa kajian risiko dalam bentuk tabel untuk setiap tahapan dalam validasi pembersihan sehingga akan ada tindakan pencegahan dan pengendalian untuk risiko dengan kategori mayor dan kritis.
Untuk memastikan keamanan, kualitas, dan efikasi obat, validasi pembersihan merupakan komponen penting dalam industri farmasi. Tujuan utama dari proses ini adalah untuk membuktikan secara terdokumentasi bahwa prosedur pembersihan yang diterapkan pada peralatan produksi dapat secara konsisten menghilangkan sisa-sisa produk sebelumnya, agen pembersih (deterjen), dan kontaminan mikroba hingga batas tertentu, sehingga aman untuk produksi selanjutnya. Kegagalan validasi pembersihan dapat menyebabkan kontaminasi silang produk, risiko kesehatan pasien, dan pelanggaran regulasi. Hal tersebut dapat menyebabkan produk ditarik dari pasar dan rusaknya reputasi perusahaan. Oleh karena itu, terdapat Good Manufacturing Practices (GMP), Cara Pembuatan Obat yang Baik (CPOB), dan APIC yang memberikan pemahaman dan penerapan prinsip validasi pembersihan (9,12,13).
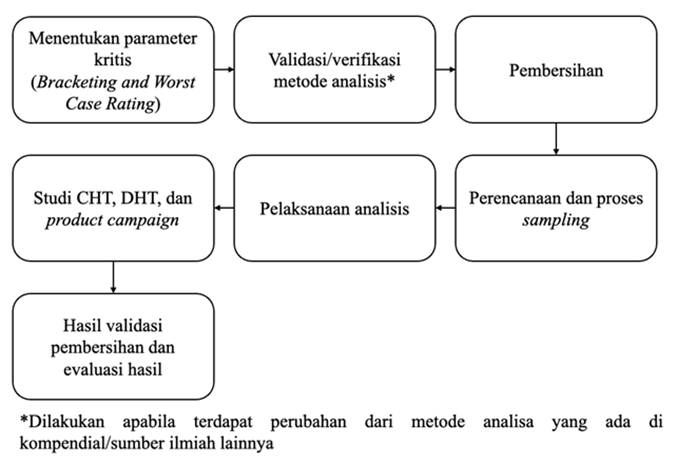
Gambar 1. Tahapan validasi pembersihan
Tahapan dalam validasi pembersihan terdapat pada Gambar 1, diawali dengan menentukan parameter kritis sehingga didapatkan zat aktif yang akan digunakan untuk validasi pembersihan. Tahapan ini penting untuk mengidentifikasi potensi bahaya dan menentukan strategi validasi, termasuk pemilihan produk dan peralatan yang dipakai atau sekiranya sulit untuk dibersihkan. Selanjutnya, batas keberterimaan residu (MACO) dihitung secara ilmiah, diikuti dengan pengembangan prosedur pembersihan yang rinci. Kemudian, ditentukan metode pengambilan sampel (swab atau bilasan) dan metode analisis (spesifik seperti HPLC dan LC/MS atau non-spesifik seperti TOC dan konduktivitas) dikembangkan dan divalidasi untuk memastikan akurasi dan recovery sampel yang memenuhi kriteria. Setelah persiapan, sampling dilakukan baik dalam pembersihan, Clean Holding Time (CHT), Dirty Holding Time (DHT), dan product campaign. Dilakukan evaluasi data dan penyusunan laporan validasi yang komprehensif untuk memastikan semua kriteria terpenuhi dan menjamin keamanan serta kualitas produk dari hasil analisa (14).
Penilaian dilakukan dengan mengamati alur selama proses validasi pembersihan seperti yang tertera pada Gambar 1. Penilaian risiko dilakukan melalui beberapa tahap sebagai berikut (15):
Identifikasi risiko dengan menggunakan metode Fishbone Ishikawa
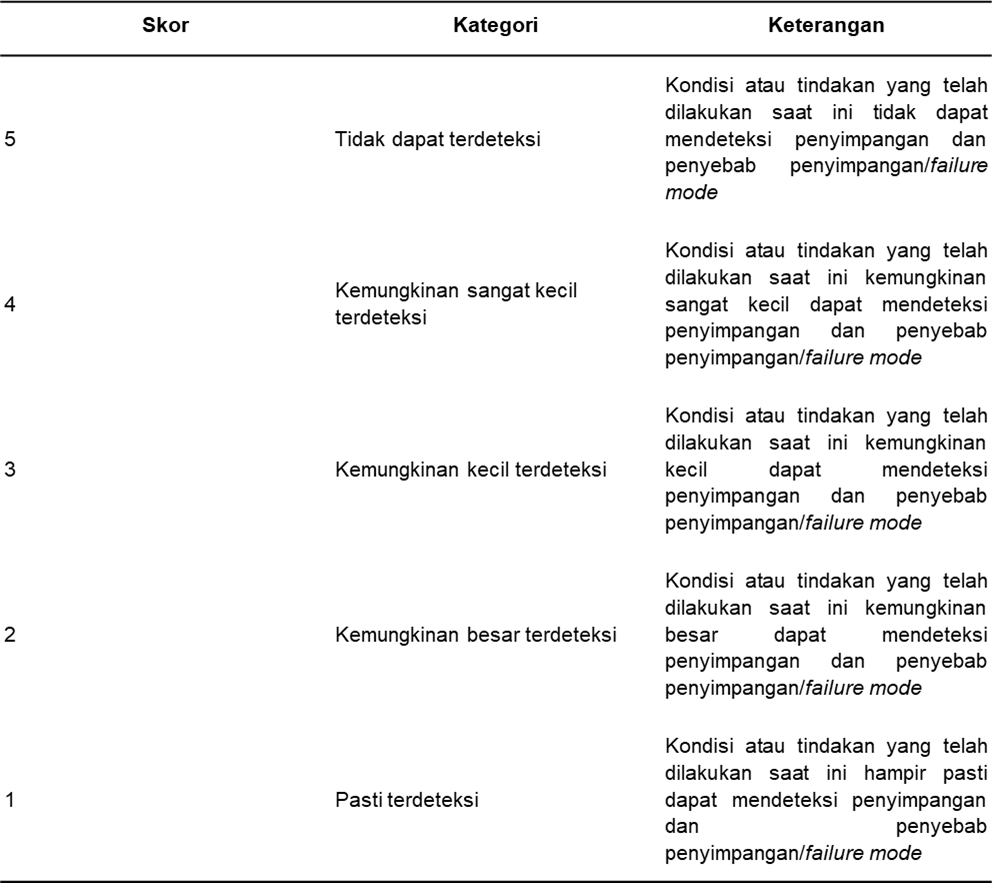
2. Analisis risiko dengan memberikan skor berdasarkan tingkat keparahan, probabilitas, dan deteksi. Kategori untuk tingkat keparahan tertera pada Tabel 1, 2, dan 3.
Tabel 1. Kategori Tingkat Keparahan (S)


Penilaian probabilitas berdasarkan tingkat kemungkinan risiko tersebut dapat terjadi dengan kategori seperti pada Tabel 2.
Tabel 2. Kategori Tingkat Probabilitas (O)


Deteksi dapat dinilai dengan menentukan kontrol yang ada sudah dapat mendeteksi dan mengantisipasi risiko yang dapat terjadi dengan kategori seperti pada Tabel 3.
Tabel 3. Kategori Tingkat Deteksi (D)
3. Evaluasi risiko dengan menghitung Risk Priority Number (RPN) untuk mendapatkan risiko yang perlu untuk dilakukan evaluasi. RPN diperoleh dengan rumus RPN = Kejadian x Probabilitas x Deteksi. Nilai RPN yang dihasilkan dapat diklasifikasikan berdasarkan kriteria seperti pada Tabel 4.
Tabel 4. Kategori Risiko Berdasarkan RPN

4. Membuat tindakan yang direkomendasikan dan menghitung nilai RPN yang baru bagi risiko yang masuk ke dalam kategori mayor sampai kritis
Hasil
Hasil penilaian risiko dilakukan pada setiap tahap validasi pembersihan dimulai dari menentukan parameter kritis, validasi metode analisa, proses pembersihan, perencanaan sampling dan sampling, analisa, CHT, DHT, dan product campaign.
Tabel 5. Risiko Saat Menentukan Parameter Kritis

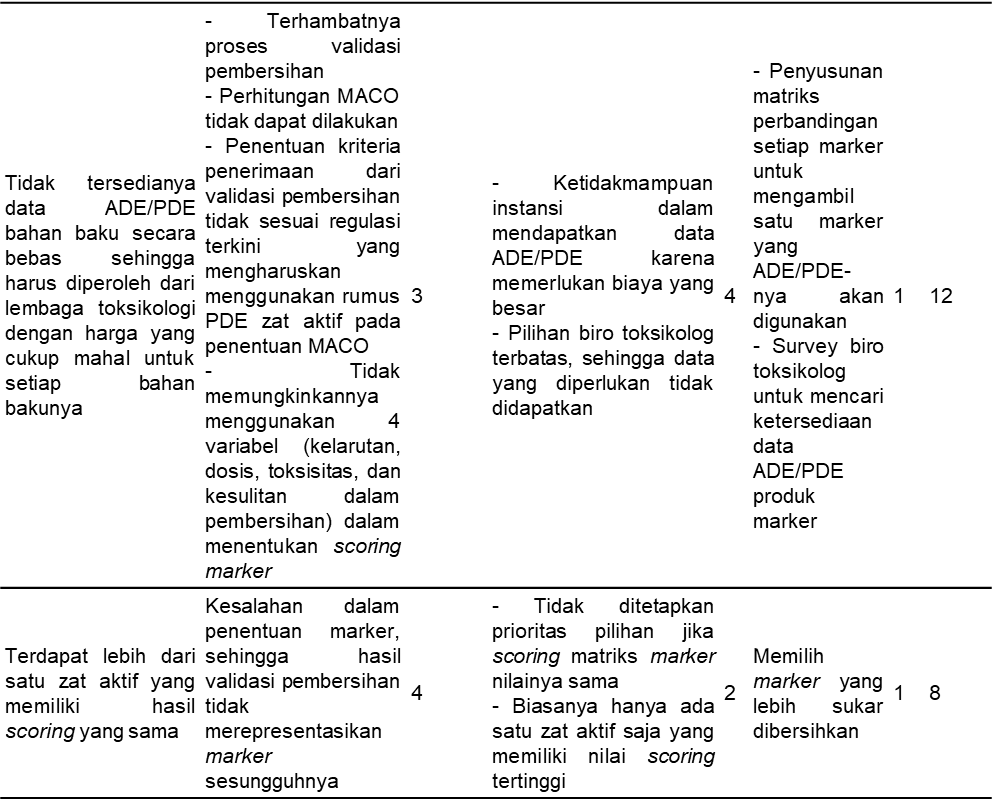
Berdasarkan hasil identifikasi dan analisis risiko, didapatkan tiga risiko pada proses menentukan parameter kritis (Tabel 5) dengan dua risiko kategori minor dan satu risiko kategori mayor yang memerlukan tindakan perbaikan.
Tabel 6. Risiko Saat Validasi/Verifikasi Metode Analisa

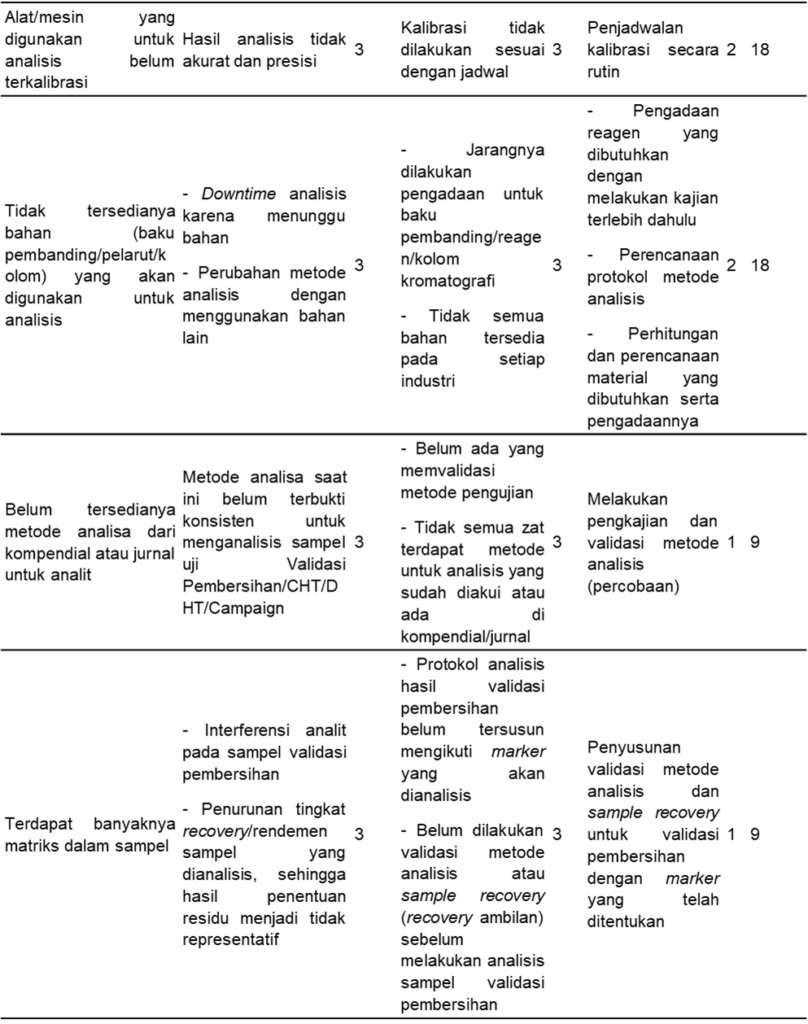
Berdasarkan hasil identifikasi dan analisis risiko, ditemukan lima risiko pada proses menentukan validasi/verifikasi metode analisa (Tabel 6) dengan dua risiko kategori minor dan tiga risiko kategori moderat.
Tabel 7. Risiko Saat Proses Pembersihan
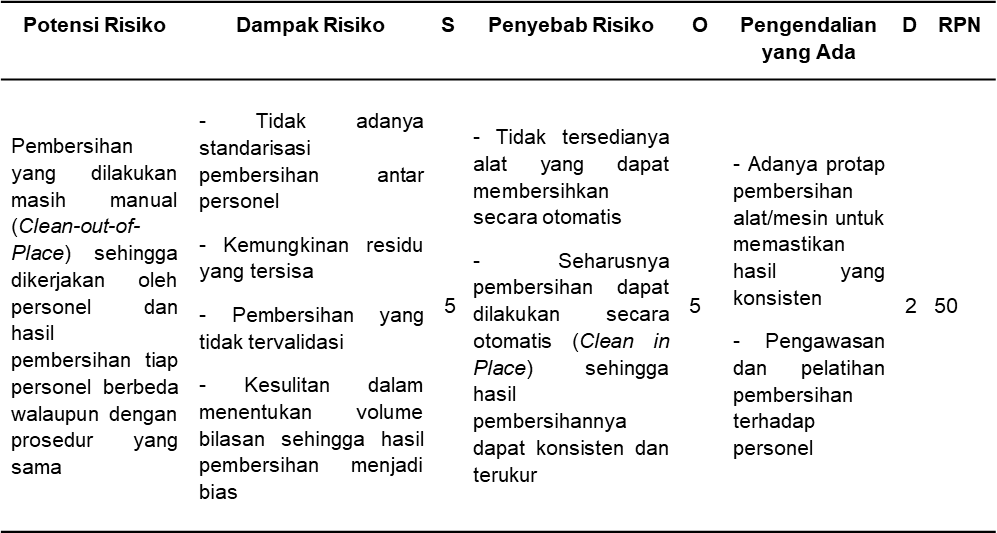
Berdasarkan hasil identifikasi dan analisis risiko, ditemukan satu risiko pada proses pembersihan (Tabel 7) dengan kategori risiko kategori mayor.
Tabel 8. Risiko Saat Sampling Validasi Pembersihan


Berdasarkan hasil identifikasi dan analisis risiko, ditemukan delapan risiko pada proses sampling validasi pembersihan (Tabel 8) dengan empat risiko kategori minor, satu risiko kategori moderat, dan tiga risiko kategori mayor
Tabel 9. Risiko Saat Proses Analisa
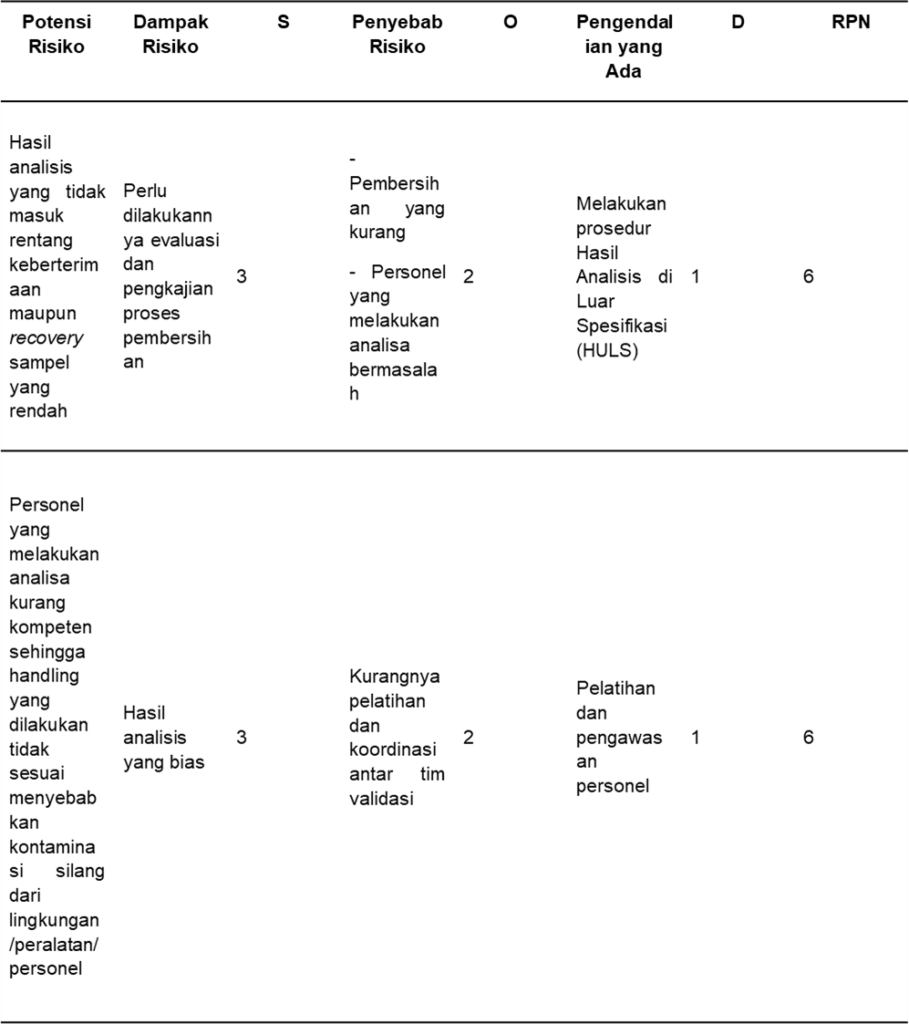
Berdasarkan hasil identifikasi dan analisis risiko, ditemukan dua risiko pada proses analisa (Tabel 9) dengan dua risiko kategori minor.
Tabel 10. Risiko Saat Studi CHT dan DHT
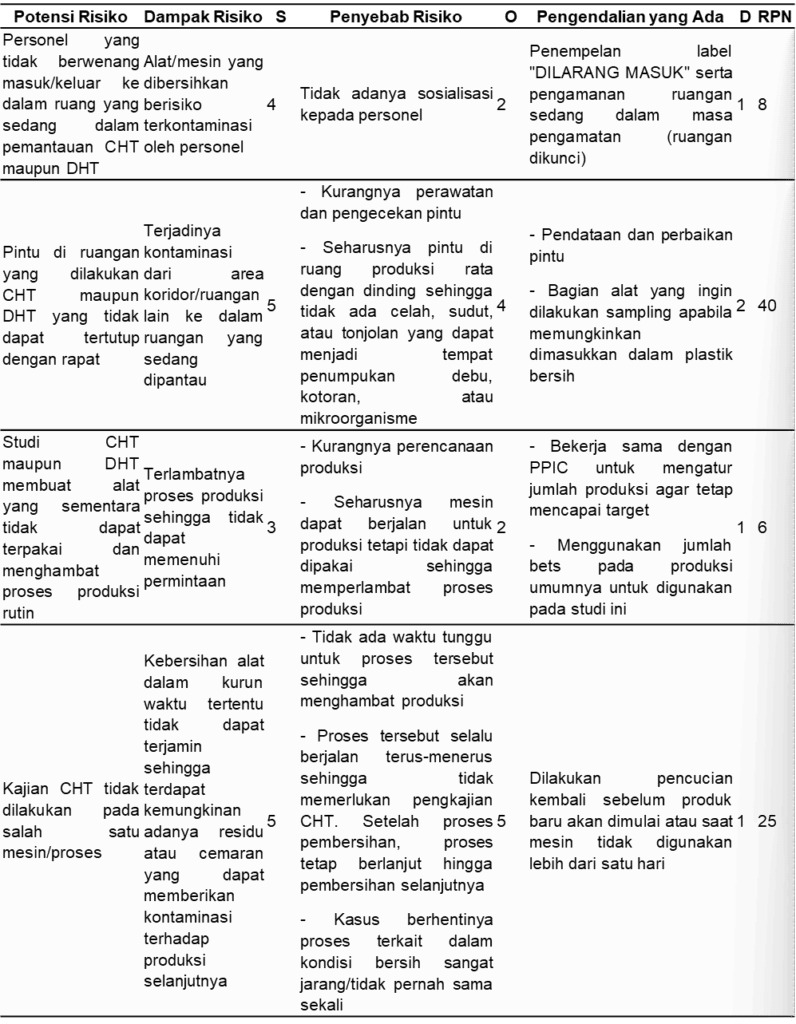

DHT (Tabel 10) dengan tiga risiko kategori minor, satu risiko kategori moderat, dan satu risiko mayor.
Tabel 11. Risiko Saat Studi Product Campaign
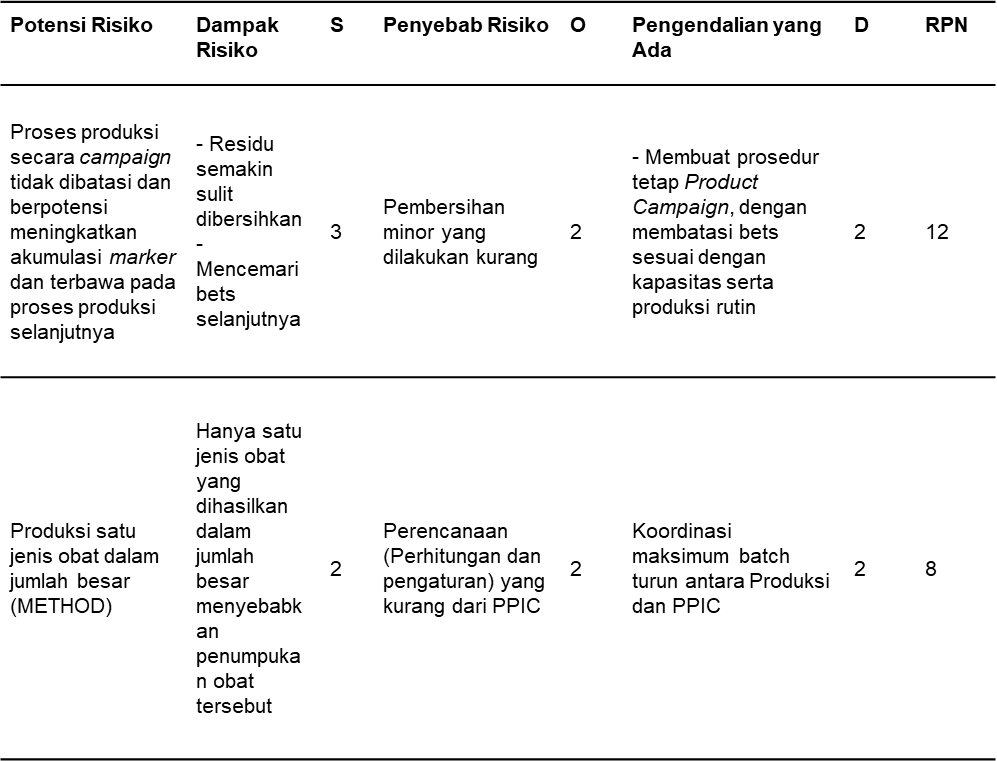
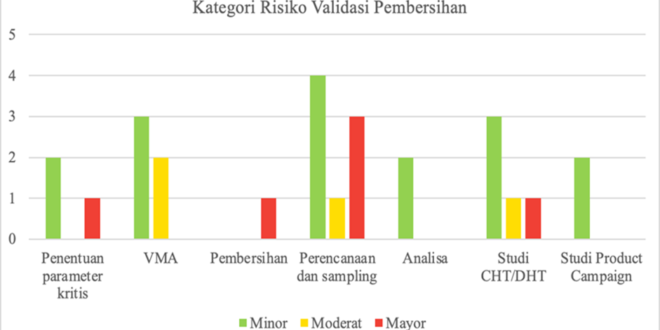
Berdasarkan hasil identifikasi dan analisis risiko, ditemukan dua risiko pada proses studi product campaign (Tabel 11) dengan kategori risiko kategori minor. Berdasarkan Tabel 5-11, didapatkan sebanyak 26 risiko dari keseluruhan proses produksi. Tahap selanjutnya didapatkan grafik risiko kontaminasi silang berdasarkan kategorinya pada setiap proses produksi seperti tercantum dalam Gambar 3.
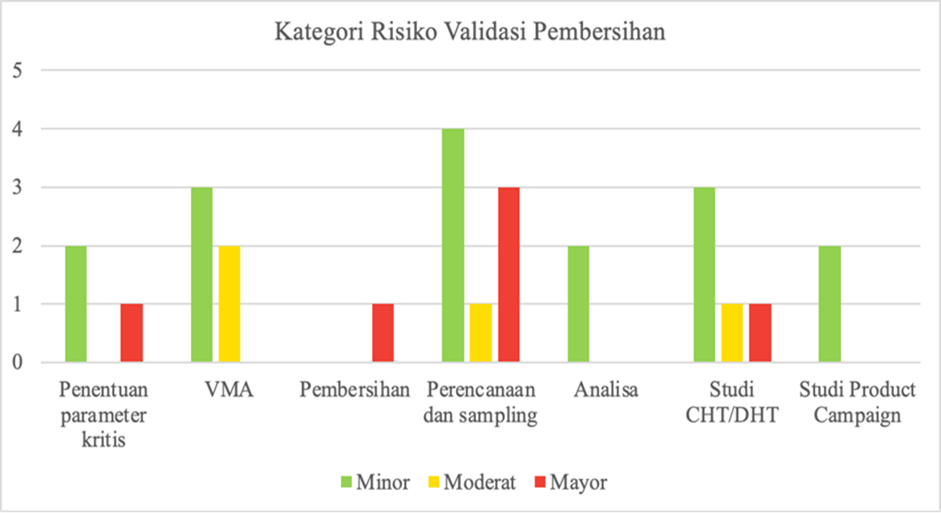
Gambar 3. Grafik kategori risiko yang dapat terjadi pada setiap proses saat validasi pembersihan
Dari hasil identifikasi risiko didapatkan sebanyak 16 risiko termasuk dalam kategori minor, 4 risiko termasuk dalam kategori moderat, dan 6 risiko termasuk dalam kategori mayor. Diperlukan evaluasi dan perhitungan RPN yang baru dari risiko mayor tersebut. Hasil evaluasi dan RPN yang baru terdapat pada Tabel 12. Evaluasi dari risiko dengan menggunakan Corrective Action and Preventive Action (CAPA) sehingga dapat mengurangi peluang terjadinya risiko serta lebih mudah dalam mendeteksi risiko.
Tabel 12. Evaluasi dan CAPA dari Risiko Dengan Kategori Mayor


Pembahasan
Dari hasil kajian risiko didapatkan beberapa hal penting yaitu risiko yang perlu ditangani maupun justifikasi terhadap kemungkinan penyimpangan yang dapat terjadi. Penentuan marker menjadi salah satu hal yang penting dalam validasi pembersihan. Pendekatan Bracketing dilakukan untuk meminimalisasi jumlah validasi yang diperlukan dengan cara mengelompokkan produk-produk terhadap serangkaian proses peralatan yang sama. Hal ini dilakukan untuk meminimalisasi waktu, tenaga, dan biaya yang dibutuhkan untuk validasi pembersihan. Tujuannya adalah sebagai alasan ilmiah untuk peringkat worst case produk dalam validasi pembersihan. Tahapannya dimulai dengan pengelompokan serangkaian peralatan yang digunakan untuk memproduksi beberapa produk (Equipment Train), yaitu yang memiliki peralatan yang sama dengan beberapa cara atau metode pembersihan yang sama. Selanjutnya, dibuat matriks mengenai produk-produk yang diproduksi berdasarkan tingkat kemudahan dan kesulitan saat proses pembersihan (Cleanability), tingkat kelarutan, nilai ADE (Acceptable Daily Exposure)/PDE (Permitted Daily Exposure), nilai toksisitas, dan dosis terapetik. Dari matriks tersebut maka akan didapatkan total nilai terbesar yang merupakan marker untuk validasi pembersihan (13,14). Marker yang dipilih harus dapat merepresentasikan untuk produk lainnya berdasarkan tingkat kesulitan dalam pembersihan, kelarutan, dosis, dan data toksikologi. Apabila terdapat marker dari produk lain yang lebih poten dari marker sebelumnya maka hasil validasi pembersihan tidak valid dan diperlukan pengulangan dengan marker yang tepat (16). Oleh karena itu, diperlukan perhitungan dan analisis yang tepat ketika melakukan pemilihan marker. Selain itu, penentuan titik kritis dan peralatan yang digunakan menjadi perhatian utama dalam tahapan ini. Akan lebih baik apabila dibuat dokumen baku tertulis yang mengatur dan mendata peralatan dan jalur yang digunakan untuk produksi masing-masing produk sehingga menjadi lebih mudah dalam menganalisisnya.
Pembersihan yang dilakukan dalam beberapa alat produksi cenderung masih dilakukan secara manual. Hal ini dikarenakan memang dalam beberapa mesin seperti mesin cetak dan mesin kemas primer belum terdapat alat yang dapat membersihkan otomatis saat ini di dunia. Namun, alat-alat untuk pencampuran/granulasi sudah ada yang dapat membersihkan otomatis walaupun tidak semua industri memilikinya (17).
Tentunya terdapat risiko dari pembersihan yang masih manual seperti tidak ada parameter yang pasti, kemungkinan hasil pembersihan yang berbeda setiap saat, dan tidak dapat mengukur volume bilasan. Untuk mengatasi risiko tersebut maka diperlukan beberapa hal seperti pelatihan terhadap personel, pembuatan prosedur pembersihan dengan suatu satuan yang dapat terukur seperti keterulangan jumlah pembersihan, jumlah cairan pembersih, serta penentuan volume bilasan. Prosedur pembersihan ini dapat diuji terlebih dahulu sampai memenuhi syarat keberterimaan yang ada. Dengan adanya pencegahan tersebut, maka personel mendapatkan cara pembersihan yang sama dengan parameter yang dapat terkuantifikasi.
Risiko lainnya yang merupakan hal kecil tetapi dapat berakibat fatal adalah terlewatnya proses pencucian serta bahan dari alat yang digunakan selama validasi pembersihan. Sudah seharusnya alat selalu dibersihkan ketika akan sampling dan setelah sampling. Namun, terdapat alat yang terbuat dari bahan yang sulit dibersihkan. Oleh karena itu, diperlukan evaluasi terhadap bahan alat sampling. Apabila bahan dari alat sampling dapat diganti dengan bahan yang mudah dibersihkan seperti stainless steel (18).
Sarana penunjang kritis dalam industri farmasi seperti HVAC (Heating, Ventilation, and Air Conditioning) yang bermasalah dapat menjadi risiko terhadap validasi pembersihan. Apabila HVAC tidak baik dalam melakukan pertukaran udara maka akan mengakibatkan kontaminasi silang atau hasil pembersihan menjadi kotor kembali (19). Kualifikasi ulang dan perawatan dari HVAC menjadi salah satu cara untuk mengurangi risiko tersebut (20).
Clean Holding Time (CHT) adalah waktu maksimum yang diizinkan bagi peralatan produksi untuk tetap berada dalam kondisi bersih setelah proses pembersihan selesai dan sebelum digunakan kembali. Sedangkan Dirty Holding Time (DHT) adalah waktu maksimum yang diizinkan bagi peralatan produksi untuk tetap berada dalam kondisi kotor setelah produksi selesai dan sebelum proses pembersihan dimulai (13,14,21). Pada saat studi CHT/DHT, ruangan yang sedang diamati harus dapat tertutup rapat (21). Namun, terdapat kemungkinan bahwa terdapatnya celah pada pintu ruangan tersebut sehingga menyebabkan debu atau kotoran dapat masuk ke dalam ruangan dan mengakibatkan hasil analisis menjadi bias. Dibutuhkan tindakan korektif seperti perbaikan pada pintu dengan memberikan ganjalan berupa karet atau plastik yang tidak dapat menyimpan debu. Selain itu, tindakan pencegahan seperti perawatan dan pendataan kondisi pintu serta ruangan diperlukan untuk tahap ini.
Apabila produksi harus berjalan secara berkelanjutan dan terdapat salah satu proses produksi yang tidak pernah berhenti karena tidak ada cadangan alat, maka studi CHT/DHT dapat tidak dilakukan dengan menganalisis risiko yang ada. Apabila mesin terus dipakai maka pembersihan akan selalu dilakukan sehingga CHT/DHT tidak dibutuhkan. Memaksakan untuk studi dilakukan dapat mengakibatkan risiko yang lebih parah seperti terlambatnya produksi sehingga permintaan tidak dapat dipenuhi.
Risiko yang dapat terjadi selama proses validasi pembersihan mulai dari risiko yang memang umum terjadi dan tindakan pencegahan yang sudah umum dilakukan sampai risiko yang khusus terjadi dalam validasi pembersihan. Risiko ini perlu untuk dipertimbangkan, dianalisis, dan dikaji untuk mengurangi potensi dampak dari risiko ini di masa yang akan datang.
Kesimpulan
Dari hasil kajian risiko didapatkan sebanyak 26 risiko dengan 16 risiko minor, 4 risiko moderat dan 6 risiko mayor yang dapat terjadi saat validasi pembersihan. Risiko yang termasuk dalam kategori mayor telah dibuat tindakan pengendalian risiko menggunakan CAPA. Hasil dari CAPA ini akan mengurangi risiko terjadinya kegagalan dalam validasi pembersihan mengingat hasilnya dapat mempengaruhi mutu produk. Oleh karena itu, industri farmasi perlu mempertimbangkan beberapa hal sebelum melakukan validasi pembersihan. Hal ini meliputi pemilihan marker yang tepat, kesiapan metode analisis, metode pembersihan yang tepat, mempertimbangkan untuk pembersihan dilakukan secara otomatis, serta sarana penunjang kritis yang memadai. Selain itu, diperlukannya kalibrasi serta kualifikasi ulang terhadap instrumen yang dibutuhkan dalam validasi pembersihan untuk menjaga data yang dihasilkan tetap valid. Diharapkan kajian ini dapat menjadi pedoman bagi suatu industri farmasi sebelum melakukan validasi pembersihan.
Daftar Pustaka
1. Ferro Uriguen A, Beobide Telleria I, Martínez Arrechea S, Miró Isasi B, Sampedro Yangüela C, Urretavizcaya Anton M. Determination of the cross-contamination and validation of the cleaning process for an automated personalised dosing system. Eur J Hosp Pharm. 2022 May;29(3):157–63.
2. Tanyous JN. Cleaning Validation: Complete Guide for Health – Based Approach in Chemical Cross – Contamination Risk Assessment. PDA Journal of Pharmaceutical Science and Technology. 2019;73(2):204–10.
3. Spoerk M, Koutsamanis I, Matić J, Eder S, Patricia Alva Zúñiga C, Poms J, et al. Novel Cleaning-in-Place Strategies for Pharmaceutical Hot Melt Extrusion. Pharmaceutics. 2020 June 24;12(6):588.
4. Pant KJ, Cotter PD, Wilkinson MG, Sheehan JJ. Towards sustainable Cleaning‐in‐Place (CIP) in dairy processing: Exploring enzyme‐based approaches to cleaning in the Cheese industry. Comp Rev Food Sci Food Safe. 2023 Sept;22(5):3602–19.
5. Coelho AS, Arribada RG, Lages EB. Cleaning Validation for Residual Estimation of Mometasone Furoate on Stainless-Steel Surface of Pharmaceutical Manufacturing Equipment Using a UHPLC-UV Method. PDA Journal of Pharmaceutical Science and Technology. 2020;74(1):41–8.
6. Bambang Gunawan RM. The Role Of Risk Management In Enhancing Company Resilience. Eduvest. 2024 May 25;4(5):4151–9.
7. Pascarella G, Rossi M, Montella E, Capasso A, De Feo G, Botti G, et al. Risk Analysis in Healthcare Organizations: Methodological Framework and Critical Variables. RMHP. 2021 July;Volume 14:2897–911.
8. ICH. Quality Risk Management Q9. ICH Harmonised Tripartite Guideline; 2005.
9. BPOM. Peraturan BPOM No. 25 Tahun 2025 tentang Cara Distribusi Obat yang Baik. BPK RI; 2025.
10. El-Awady SMM. Overview of Failure Mode and Effects Analysis (FMEA): A Patient Safety Tool. Global Journal on Quality and Safety in Healthcare. 2023 Feb 1;6(1):24–6.
11. Van Der Galiën R, Langen AL, Jacobs LJM, Hagen B, Flahive K, Chatterjee SD, et al. Setup of a Contamination Control Strategy Using the Hazard Analysis Critical Control Point (HACCP) Methodology. PDA Journal of Pharmaceutical Science and Technology. 2023;77(4):317–28.
12. Valavala S, Seelam N, Tondepu S, Sundaramurthy V. Cleaning Method Validation for Estimation of Dipyridamole Residue on the Surface of Drug Product Manufacturing Equipment Using Swab Sampling and by High Performance Liquid Chromatographic Technique. tjps. 2020 Apr 1;17(2):182–9.
13. APIC. Guidance on Aspect Of Cleaning Validation in Active Pharmaceutical Ingredient Plants. Active Pharmaceutical Ingredients Committee; 2021.
14. Singh K, Tamta B, Mukopadayay S. Cleaning validation process in pharmaceutical industry: A review. ijhs. 2022 June 7;13557–73.
15. McGowan J, Wojahn A, Nicolini JR. Risk Management Event Evaluation and Responsibilities. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 [cited 2025 Oct 13]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK559326/
16. Moura MJ, Pereira AD, Santos DJF, Silva AG, Paiva CCAD, Duarte BPM. Cleaning validation in pharmaceutical quality control laboratories: a structured protocol for contamination risk mitigation. DARU J Pharm Sci. 2025 July 3;33(2):20.
17. Röcker R, Eggers B, Kramer A, Stope MB. Effective manual cleaning as the first step of reprocessing glass probes of a medical device for non-invasive physical plasma therapy. GMS Hygiene and Infection Control [Internet]. 2024 [cited 2025 Oct 14];19. Available from: https://journals.publisso.de/en/journals/hic/volume19/dgkh000486
18. Benčina M, Rawat N, Paul D, Kovač J, Iglič A, Junkar I. Surface Modification of Stainless Steel for Enhanced Antibacterial Activity. ACS Omega. 2025 Apr 8;10(13):13361–9.
19. Tune Tibesso D, Gabriel T, Balcha Balla T, Belete A. Compliance of Pharmaceutical Manufacturing Companies to Good Manufacturing Practices in Heating, Ventilation, and Air‐Conditioning Systems: The Case of Local Ethiopian Firms. Natalini B, editor. Advances in Pharmacological and Pharmaceutical Sciences. 2024 Jan;2024(1):6109415.
20. Dhandapani K, Kella A, Narayanasamy D. Maintaining a Sterile Environment: Validation and Qualification Strategies for Heating, Ventilation, and Air Conditioning Systems Adhering to Current Good Manufacturing Practices in Pharmaceutical Facilities. Cureus [Internet]. 2024 Sept 1 [cited 2025 Oct 13]; Available from: https://www.cureus.com/articles/279435-maintaining-a-sterile-environment-validation-and-qualification-strategies-for-heating-ventilation-and-air-conditioning-systems-adhering-to-current-good-manufacturing-practices-in-pharmaceutical-facilities
21. Patera J, Štípková G, Zámostný P, Bělohlav Z, Vltavský Z. Effect of dirty-hold time on cleaning process of pharmaceutical equipment. Pharmaceutical Development and Technology. 2013 Feb;18(1):274–9.
cara mengutip artikel
https://jurnal.unpad.ac.id/farmasetika/rt/captureCite/67440/26661