Majalah Farmasetika Jurnal Ilmiah Nasional Terakreditasi SINTA 3
Majalah Farmasetika Jurnal Ilmiah Nasional Terakreditasi SINTA 3
Artikel Terkait

Majalah Farmasetika, 9 (4) 2024, 351-366
https://doi.org/10.24198/mfarmasetika. 9i4.56576
Artikel Review
Viviane Annisa*
Departemen Farmasi, Fakultas Matematika dan Ilmu Pengetahuan Alam, Universitas Islam Indonesia, Yogyakarta
*E-mail : viviane@uii.ac.id
(Submit 24/07/2024, Revisi 29/07/2024, Diterima 03/08/2024, Terbit 13/08/2024)
Abstrak
Good Manufacturing Practice (GMP) merupakan salah satu golden standar dari pemerintah untuk menilai kesesuaian proses produksi dan keamanan produk. Prinsip dari GMP adalah kualitas dibangun kedalam produk, tidak hanya pada pengujian saja. Pembuatan obat di Indonesia mengacu pada standar GMP yang berlaku di Indonesia untuk industri farmasi, yakni Pedoman CPOB. Kajian ini betujuan untuk melakukan kajian tentang penerapan GMP di industri farmasi Indonesia, sehingga dapat memberikan gambaran bagaimana sistem GMP diterapkan di Indonesia. Metode penulisan artikel menggunakan berbagai referensi dari pedoman standar praktik atau GMP di Indonesia dan artikel publikasi. Dari hasil studi ini ditemukan bahwa topik pembahasan yang terkait industri farmasi, meliputi tentang sarana penunjang berupa tata udara ruangan, pengolahan limbah, bangunan seperti area penyimpanan/gudang dan lantai, analisis kapabilitas dan kinerja mesin, analisis risiko, dan Keamanan Dan Keselamatan Kerja (K3). Terdapat beberapa aspek GMP untuk industri farmasi obat yang harus dipenuhi, antara lain: sistem mutu produk, personalia, bangunan dan fasilitas, peralatan, produksi, cara penyimpanan dan pengiriman, pengawasan mutu, inspeksi, audit mutu, audit, dan persetujuan pemasok, keluhan dan penarikan produk, dokumentasi, kegiatan alih daya, kualifikasi dan validasi. Manfaat dari kepatuhan industri farmasi terhadap GMP adalah dapat memproduksi produk secara konsisten, sehingga dapat mengurangi risiko proses produksi atau menurunkan angka produk defect yang nantinya akan berpengaruh terhadap pertumbuhan profit industri farmasi. Kesimpulan studi ini adalah kepatuhan terhadap GMP penting diperhatikan agar obat yang diproduksi oleh industri farmasi terjamin mutu, keamanan, dan efektifivitasnya.
Kata kunci: OB, GMP, industri farmasi, BPOM, FDA
Teks Lengkap:
Pendahuluan
Good Manufacturing Practice (GMP) merupakan salah satu golden standar dari pemerintah untuk menilai kesesuaian proses produksi dan keamanan produk (1). Menurut WHO, GMP merupakan bagian dari pemastian mutu yang memastikan produk diproduksi secara konsisten dan dikontrol agar memenuhi standar kualitas yang berlaku sesuai dengan tujuan penggunaannya dan persyaratan izin edar. Prinsip dari GMP adalah kualitas dibangun kedalam produk, tidak hanya pada pengujian saja (2). Setiap pemerintah di masing-masing negara, khususnya di negara berkembang, mengalokasikan proporsi untuk obat-obatan lebih dari 40%. Maka, pentingnya memastikan kualitas produksi pada industri farmasi, agar obat yang diproduksi sesuai dengan standar kualitas, keamanan, dan efikasi. Banyak artikel yang membahas tentang hubungan antar produk obat dan pasien. Obat yang berkualitas dapat berpengaruh langsung terhadap efektivitas pengobatan pasien (3).
Dalam konteks komersial, interaksi antara konsumen dan pelaku usaha diatur oleh perjanjian yang mengikat secara hukum. Persyaratan hukum perjanjian dan asas-asas perjanjian merupakan aspek yang paling signifikan dalam hukum perjanjian yang berkaitan dengan hubungan antara pelaku usaha dan konsumen (4). Aturan mengenai perlindungan konsumen telah diatur dalam UU Perlindungan Konsumen No. 8 Tahun 1999, yang mencakup segala upaya yang menjamin kepastian hukum untuk memberi perlindungan kepada konsumen. Konsumen memiliki hak atas kenyamanan, keaman, dan keselamatan dalam mengkonsumsi barang, sehingga kewajiban produsen adalah menjamin mutu barang yang diproduksi berdasarkan ketentuan standar mutu barang yang berlaku (5).
Dalam hal ini, pembuatan obat di Indonesia mengacu pada standar GMP yang berlaku di Indonesia untuk industri farmasi, yakni Pedoman CPOB Tahun 2018 (Cara Pembuatan Obat yang Baik) yang diatur oleh Badan Pengawas Obat dan Makanan (BPOM) (6). Menurut UU No. 1799/MENKES/PER/XII/2010 tentang Industri Farmasi, CPOB adalah cara pembuatan obat yang bertujuan untuk memastikan agar mutu obat yang dihasilkan sesuai dengan persyaratan dan tujuan penggunaannya. Pembuatan obat harus memenuhi persyaratan CPOB yang dibuktikan dengan sertifikat CPOB (berlaku selama 5 tahun sepanjang memenuhi persyaratan) (7). Pedoman CPOB ini mengacu pada WHO TRS 981 Tahun 2012 (Annex 2); WHO TRS 986 Tahun 2013 (Annex 5); WHO TRS 992 Tahun 2014 (Annex 3 dan Annex 5); WHO TRS 996 (Annex 5) Tahun 2015; WHO TRS 999 Tahun 2016 (Annex 2). Cakupan CPOB meliputi seluruh aspek produksi dan pengendalian mutu. CPOB menjadi acuan yang wajib diterapkan di industri farmasi yang melakukan kegiatan pembuatan obat dan bahan obat (6).
Kajian tentang penerapan GMP penting dilakukan, karena dapat memberikan gambaran mengenai penerapan GMP di industri farmasi Indonesia. Dengan adanya gambaran mengenai penerapan GMP, maka apoteker industri farmasi agar dapat mengevaluasi tingkat kepatuhan industri farmasi terhadap GMP, khususnya untuk industri farmasi obat mengacu pada CPOB dan senantiasa berorientasi terhadap mutu, keamanan, dan efektivitas obat yang diproduksi. Artikel publikasi yang membahas tentang GMP di Indonesia masih belum ada, sehingga nilai kebaruaannya tinggi. Maka dari itu, pada artikel ini dilakukan review artikel dari publikasi-publikasi terkait serta penerapannya di industri farmasi Indonesia dan secara khusus membahas tentang Overall Equipment Effectiveness (OEE) dan Kualifikasi Sistem Tata Udara pada industri farmasi.
Metode
Metode penulisan artikel menggunakan berbagai referensi dari pedoman standar praktik atau GMP di Indonesia dan artikel dengan kata kunci “CPOB” dan “Industri”, dan “Farmasi” dari database Google Scholar dengan rentang tahun 2014-2024 menggunakan jenis artikel dan review artikel. Artikel yang diperoleh sebanyak 952 dokumen, kemudian disaring hanya artikel publikasi saja. Lalu disaring lagi sesuai dengan kriteria inklusi dan eksklusi. Kriteria inklusi meliputi: artikel tentang penerapan CPOB sesuai aspeknya di industri farmasi dan terkait dengan bidang farmasi. Kriteria eksklusi meliputi: artikel yang tidak terkait dengan bidang farmasi, seperti analisa ekonomi, keuangan perusahaan, marketing, hukum, pelatihan, dll.
Hasil dan Pembahasan
Kajian GMP di Indonesia
Acuan GMP di Indonesia menggunakan CPOB. Komparasi aspek-aspek CPOB dengan standar GMP di negara lainnya seperti USA yang menggunakan FDA dan Eropa yang menggunakan EMA dapat dilihat di Tabel 1. Secara keseluruhan, aspek-aspek dari ketiga standar tersebut hampir sama, sehingga dapat diterapkan antar negara tersebut. Aspek-aspek GMP untuk industri farmasi secara garis besar mencakup manajemen mutu, personalia, bangunan dan fasilitas, peralatan, sanitasi dan higiene, produksi, pengawasan mutu, inspeksi diri, keluhan dan penarikan, dokumentasi, kontrak, kualifikasi, dan validasi.
Tabel 1 Komparasi GMP dari Indonesia, USA, dan Eropa (6,8,9)

A. Manajemen Mutu
Sektor farmasi harus memproduksi obat-obatan yang sesuai dengan tujuan penggunaannya, memenuhi persyaratan registrasi, dan tidak menimbulkan bahaya dalam penggunaannya karena berbahaya, berkualitas buruk, atau tidak efektif. Manajemen bertanggung jawab untuk mencapai tujuan ini melalui “Kebijakan Mutu” yang menuntut partisipasi dan komitmen dari semua departemen perusahaan, serta pemasok dan distributor (6,8,9).
B. Personalia
Sumber daya manusia sangat penting dalam pengembangan dan penerapan sistem jaminan kualitas yang efektif dan produksi obat-obatan yang tepat. Sektor farmasi harus memiliki staf yang berkualitas dan berpengalaman dalam jumlah yang memadai, serta kerangka kerja organisasi. Kepala bagian produksi, kepala bagian pengawasan mutu dan kepala bagian manajemen mutu merupakan tokoh-tokoh penting dalam sektor farmasi. Ketiga orang kunci tersebut harus independen satu sama lain (6,8,9).
C. Bangunan dan Fasilitas
Bangunan dan fasilitas yang digunakan untuk pembuatan obat harus memiliki desain, konstruksi, dan lokasi yang sesuai, serta dikondisikan dan dipelihara dengan baik untuk memastikan fungsinya dengan baik. Tata letak dan desain ruangan harus sedemikian rupa sehingga meminimalkan risiko kesalahan, kontaminasi silang, dan kesalahan lainnya, serta memfasilitasi pembersihan, sanitasi, dan pemeliharaan yang efektif untuk menghindari kontaminasi silang, penumpukan debu atau kotoran, dan efek lain yang dapat mengurangi kualitas obat (6,8,9).
D. Peralatan
Peralatan untuk pembuatan obat harus memiliki desain dan konstruksi yang tepat, ukuran yang memadai, serta lokasi yang tepat dan memenuhi syarat untuk memastikan kualitas obat sesuai dengan desain dan seragam dari satu bets ke bets lainnya, serta untuk memudahkan pembersihan dan perawatan guna mencegah kontaminasi silang, penumpukan debu atau kotoran, dan masalah-masalah lain yang secara umum berdampak negatif terhadap kualitas produk (6,8,9).
E. Sanitasi dan Higiene
Setiap elemen dalam pembuatan obat harus tunduk pada standar sanitasi dan higiene yang ketat. Personil, bangunan, peralatan, dan perlengkapan, bahan dan wadah produksi, bahan pembersih dan desinfeksi, serta segala sesuatu yang dapat menyebabkan kontaminasi produk, semuanya tercakup dalam sanitasi dan higiene. Program sanitasi dan higiene yang lengkap dan terintegrasi harus diterapkan untuk menghilangkan potensi kontaminan (6,8,9).
F. Produksi
Produksi hendaklah dilaksanakan dengan mengikuti prosedur yang telah ditetapkan, dan memenuhi ketentuan CPOB yang menjamin produk selalu memenuhi persyaratan mutu serta memenuhi ketentuan izin pembuatan dan izin edar. Produksi hendaklah dilakukan dan diawasi oleh personil yang kompeten. Produk dan bahan hendaklah dilindungi terhadap pencemaran mikroba atau pencemaran lain pada tiap tahap pengolahan (6,8,9).
G. Pengawasan Mutu
Produksi harus dilakukan sesuai dengan peraturan CPOB dan protocol yang telah ditetapkan untuk menjamin bahwa produk akhir selalu memenuhi standar kualitas dan persyaratan perizinan baik untuk pembuatan maupun distribusi. Tenaga profesional dengan keahlian yang diperlukan harus mengawasi dan mengelola produksi. Pada setiap tahap pengolahan, bahan dan produk harus dilindungi dari kontaminasi mikroba atau kontaminasi lainnya (6,8,9).
H. Inspeksi Diri
Mengevaluasi apakah setiap aspek pengawasan mutu dan produksi di sektor farmasi telah memenuhi standar CPOB adalah tujuan dari inspeksi diri. Tujuan dari program inspeksi diri adalah untuk mengidentifikasi setiap kekurangan dalam penerapan CPOB dan menentukan apa yang harus dilakukan untuk memperbaikinya. Inspeksi diri harus dilakukan secara menyeluruh dan independen oleh karyawan yang berkualifikasi dari perusahaan yang mampu menilai penerapan CPOB secara tidak memihak. Inspeksi rutin juga harus dilakukan dalam keadaan tertentu, seperti ketika obat yang sudah jadi ditarik kembali atau ketika penolakan terjadi berulang kali. Setiap rekomendasi untuk tindakan perbaikan harus dilaksanakan. Program tindak lanjut yang berhasil harus dilaksanakan, dan protokol serta catatan inspeksi diri harus dipelihara. Tujuan dari inspeksi diri adalah untuk menilai seberapa baik pabrik mematuhi GMP dalam hal QC dan produksi. Tujuan dari inspeksi mandiri adalah untuk mengidentifikasi masalah yang muncul selama penerapan GMP. Inspeksi diri dilakukan secara teratur dan sesuai kebutuhan. Personil yang memenuhi syarat untuk menilai penerapan GMP secara obyektif harus ada dalam tim yang relevan. Personil yang kompeten secara independen melakukan inspeksi mandiri. Setiap temuan dari inspeksi diri dicatat, bersama dengan rekomendasi untuk tindakan perbaikan dan pengamatan yang dilakukan selama pemeriksaan. Diperlukan kampanye tindak lanjut yang efisien. Untuk memastikan kepatuhan terhadap standar jaminan mutu, aspek inspeksi diri harus ditinjau secara berkala sesuai dengan program yang telah ditetapkan. WHO diacu dalam aspek inspeksi diri CPOB yang berkaitan dengan personalia, bangunan, termasuk fasilitas personalia, pemeliharaan bangunan dan peralatan, penyimpanan bahan baku dan barang jadi, peralatan, produksi dan IPC, pengawasan mutu, dokumentasi, sanitasi dan higiene, program validasi dan revalidasi, dan kalibrasi instrumen atau sistem pengukuran, prosedur penarikan kembali, pengelolaan keluhan, pengawasan label, hasil inspeksi diri sebelumnya dan tindakan perbaikan (6,8,9).
I. Keluhan dan Penarikan
Semua keluhan dan bahan lain yang berkaitan dengan potensi cacat obat harus ditinjau secara menyeluruh sesuai dengan protokol yang ditetapkan. Untuk menangani semua kasus yang mendesak, sebuah prosedur harus dibuat, yang mungkin melibatkan penarikan produk yang diketahui atau dicurigai cacat dari peredaran secara tepat waktu dan efektif (6,8,9).
J. Dokumentasi
Dokumentasi adalah komponen dari sistem informasi manajemen, dan dokumentasi yang memadai sangat penting untuk jaminan kualitas. Dokumentasi yang jelas sangat penting untuk memastikan bahwa tiga pekerja menerima deskripsi yang jelas dan terperinci tentang tugas-tugas penting, sehingga mengurangi kemungkinan salah tafsir dan kesalahan yang biasanya terjadi jika hanya mengandalkan komunikasi lisan (6,8,9).
K. Kontrak
Pembuatan dan analisis kontrak harus dibuat, disetujui, dan dikendalikan dengan baik untuk menghindari kesalahpahaman yang dapat menyebabkan kualitas produk atau pekerjaan yang tidak memuaskan. Kontrak tertulis antara Kontraktor dan Penerima Kontrak harus dibuat dengan jelas yang menetapkan tanggung jawab dan kewajiban masing-masing pihak. Kontrak tersebut harus secara jelas menyatakan prosedur untuk meloloskan setiap batch produk untuk diedarkan yang merupakan tanggung jawab penuh dari kepala Jaminan Mutu (6,8,9).
L. Kualifikasi dan Validasi
CPOB memberikan mandat kepada sektor farmasi untuk menentukan validasi yang harus dilakukan sebagai bukti kontrol atas komponen penting dari operasi yang dilakukan. Validasi setiap modifikasi yang signifikan pada fasilitas, peralatan, atau prosedur yang mungkin berdampak pada kualitas produk. Cakupan dan luasnya validasi harus ditentukan dengan menggunakan metodologi penilaian risiko (6,8,9).
Hasil Literatur Review
Artikel publikasi yang diperoleh tentang Good Manucfacturing Practice (GMP) industri farmasi di Indonesia dapat dilihat pada Tabel 2. Topik pembahasan yang terkait industri farmasi meliputi tentang sarana penunjang berupa tata udara ruangan, pengolahan limbah, bangunan seperti area penyimpanan/gudang dan lantai, analisis kapabilitas dan kinerja mesin, analisis risiko, dan Keamanan Dan Keselamatan Kerja (K3).
Tabel 2 Publikasi Terkait Good Manucfacturing Practice (GMP) di Indonesia

A. Overall Equipment Effectiveness (OEE)
Setiap industri pasti memiliki keinginan untuk memproduksi produk yang berkualitas dan profit yang tinggi dengan mereduksi biaya produksi. Hal ini dapat dicapai dengan mengimplementasikan sistem operasional mesin yang efektif. Peningkatan produktivitas mesin dapat meningkatkan profit industri. Mesin produksi merupakan bagian penting dalam sistem manufaktur dan efisiensinya berpengaruh langsung terhadap kualitas dan biaya dari produk dan keseluruhan produktivitas dari industri. Salah satu indikator performa yang dapat digunakkan adalah Overall Equipment Effectiveness (OEE). Instrumen OEE menghitung bagaimana keefektivan suatu operasional manufaktur. OEE telah ada sejak 50 tahun yang lalu, diciptakan oleh Siichi Nakajima dari Jepang, pioneer sistem Total Productive Maintenance (TPM) (30). Dalam TPM, performa sebuah sistem produktif diukur dengan metrik kuantitatif inti yang dikenal dengan OEE. Implementasi OEE dalam industri akan meningkatkan kualitas produk, menurunkan peralatan rusak/break down, idle time, tingkat kecelakaan, dan produk defect/reject (31).
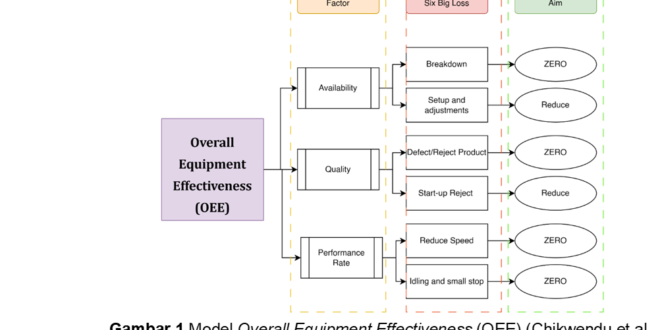
Istilah Six Big Losses dikenal sebagai enam kerugian besar yang sering ditimbulkan dari perusahaan akibat penggunaan mesin yang tidak efektif dan efisien (32). Pada Gambar 1 dapat dilihat Six Big Losses dari TPM diklasifikasikan ke dalam 6 kategori mayor, yaitu breakdown losses, setup and adjustment losses, defect and reject losses, start-up losses, reduce speed, and idling and small stoppage losses. Berdasarkan Six Big Losses, OEE dapat dihitung dengan menentukan Availability, Performance, and Quality rate (33).
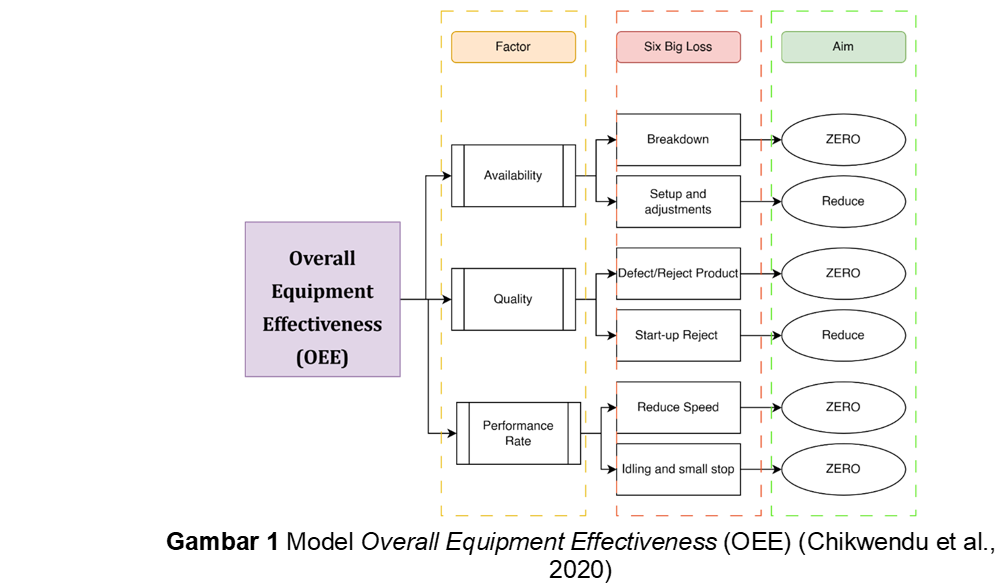
Kemudian OEE dihitung dari Availability x Performance rate x Quality rate. Standar dunia (World Class Goals) untuk setiap faktor OEE dapat dilihat pada Tabel 3. Jika nilai kalkulasi OEE ≥85%, maka dapat dikatakan industri farmasi tersebut dalam kondisi baik, sebaliknya jika ≤85% maka dapat dikatakan bahwa industri tersebut perlu ditingkatkan lagi sistem dan strategi perawatan mesinnya, jika tidak maka sulit untuk industri tersebut bertahan (31).
Tabel 3 World Class Goals untuk Overall Equipment Effectiveness (OEE) (34)

B. Kualifikasi Sistem Tata Udara
HVAC (Heating, Ventilation, and Air Conditioning) merupakan salah satu sarana penunjang kritis dalam industri Farmasi yang berperan dalam sistem pengendalian udara. Adapun yang dikendalikan oleh HVAC antara lain: suhu, kelembapan nisbi, arah pergerakan udara, jumlah partikel maupun pertukaran udara. Salah satu komponen utama dalam HVAC adalah filter yang meliputi pre filter, medium filter, dan HEPA (High-Efficiency Particulate Absorbing/Air). Pemeliharaan terhadap filter sangat penting dilakukan sebagai salah satu upaya untuk menjamin kualitas udara bersih yang dihasilkan oleh HVAC. Parameter yang menunjukkan bahwa filter harus diganti adalah dengan melihat batas maksimum yang telah ditentukan pada manometer magnehelic. Kualifikasi pada peralatan dalam industri farmasi untuk memastikan bahwa peralatan harus terpasang dengan baik sehingga dapat berfungsi sesuai dengan fungsinya. Peran penting sistem pengendalian udara dengan HVAC bertujuan untuk mencapai kelas kebersihan ruang sesuai dengan persyaratan CPOB, memberi perlindungan terhadap produk dari kontaminasi silang (cross contamination), serta memberi lingkungan kerja yang nyaman bagi karyawan. HVAC sangat menentukan kelas kebersihan ruang pada industri farmasi (6).
Sistem HVAC ini meliputi PAC (Package Air Conditioning), ducting, filter, booster fan, dumper, grille, dan panel kontrol. Perawatan AHU terdiri dari pembersihan koil pendingin, penggantian filter, dan pembersihan saluran dan sistem HVAC. Filter HVAC mengelola dan mengurangi jumlah partikel dan kuman yang mencemari udara yang masuk ke dalam ruangan. Filter yang digunakan diklasifikasikan menjadi tiga jenis: pre-filter, yang bersentuhan langsung dengan udara luar; medium filter, yang merupakan filter kedua setelah pre-filter dan menyaring udara sebelum masuk ke HEPA; dan HEPA, yang merupakan penyaring akhir udara yang masuk ke dalam ruangan. Pengecekan kondisi filter dilakukan dengan alat magnehelic. Alat ini mengukur different pressure yang dihasilkan, apabila jarum menunjukkan angka diatas batas maksimum yang telah ditentukan, maka filter harus diganti. Kualifikasi bertujuan untuk menjamin peralatan, sistem atau sarana penunjang yang digunakan sesuai dengan spesfikasi dan tujuan penggunaan yang ditentukan, agar memenuhi persyaratan CPOB, dan mempunyai cukup informasi agar dapat mengoperasikan alat secara aman dan pemeliharaan yang efektif. Kualifikasi terdiri dari 4 macam yaitu Kualifikasi Desain (KD), Kualifikasi Instalasi (KI), Kualifikasi Operasional (KO) dan Kualifikasi Kinerja (KK) yang harus dilakukan berurutan mulai dari KD, KI, KO lalu KK (6).
Kualifikasi desain (KD) digunakan untuk menjamin bahwa sistem atau peralatan yang dimaksudkan memenuhi persyaratan yang ditentukan. KD dilakukan sebelum mesin, peralatan, atau fasilitas pendukung dibuat. Kualifikasi instalasi (KI) diselesaikan setelah kualifikasi desain (KD). Proses pemeriksaan instalasi untuk memastikan bahwa semua komponen memenuhi spesifikasi dan telah dipasang dengan benar. Kualifikasi operasional dilakukan setelah kualifikasi instalasi. Kualifikasi operasional adalah proses mendokumentasikan dan memastikan bahwa komponen penyusun sistem berfungsi sebagaimana mestinya dan memenuhi spesifikasi. Kualifikasi kinerja mendokumentasikan dan memastikan bahwa komponen penyusun sistem berfungsi sebagaimana mestinya, memenuhi standar kinerja yang ditetapkan, dan menghasilkan produk yang diperlukan secara konsisten dan berkelanjutan (6).
Untuk kelas A, kelas B, dan kelas C kualifikasi kinerja (KK) dilakukan pada kondisi ruang non-operasional (tidak ada produksi) dan operational (pada saat ada produksi). Sedangkan untuk kelas D dan E hanya dilakukan pada kondisi ruang non-operasional. Ruang bersih dan sarana udara dapat dinyatakan terkualifikasi apabila hasil yang diperoleh selama 5 hari berturut-turut stabil dan memenuhi persyaratan untuk kelas A, B, C dan D. Sedangkan untuk kelas E, tidak ada ketentuan harus dilakukan berapa hari. Kualifikasi kinerja HVAC 1 mengikuti ketentuan klasifikasi ruang kelas E sehingga pemeriksaan dilakukan pada kondisi ruang non-operation, untuk waktu pemeriksaan selama 3 hari berturut-turut.
Penentuan parameter kritis dan non-kritis kualifikasi kinerja (KK) HVAC menggunakan analisis resiko semua komponen, subsistem, dan pengendalian dari sistem HVAC. Parameter kritis adalah semua parameter yang dapat berdampak langsung mempengaruhi mutu produk. Parameter kritis dalam KK antara lain suhu, kelembapan, jumlah partikel, tekanan, dan aliran udara. Masing-masing parameter memiliki persyaratan tertentu yang disesuaikan dengan klasifikasi kelas kebersihan ruang tersebut.
Untuk mencapai persyaratan tersebut, juga diperlukan tindakan tambahan seperti mengenakan pakaian kerja sesuai APD (Alat Pelindung Diri), melakukan pembersihan ruangan, dan mengurangi sumber yang menghasilkan partikel seperti: personil, mesin, dan bahan dalam ruangan. Personil yang melakukan KK harus menggunakan APD yang telah ditetapkan, yaitu jas lab, masker, penutup mata dan sandal untuk dalam ruang. Ruangan untuk KK harus dibersihkan terlebih dahulu. Pembersihan ruangan pada produksi kapsul lunak dilakukan secara berkala setiap hari oleh petugas kebersihan meskipun tidak ada produksi.
Parameter yang diukur pada kualifikasi kinerja (KK) adalah sebagai berikut:
a. Suhu dan kelembapan
Suhu dan kelembapan merupakan parameter yang sangat berkaitan, biasanya diukur bersamaan pada satu alat. Pengukuran suhu dan kelembapan menggunakan alat Graphical Data Logger (GDL) yang diletakkan pada sejumlah titik lokasi tertentu dalam ruangan. Semakin luas ruangan maka diperlukan titik sampling yang lebih banyak sehingga dibutuhkan alat GDL yang lebih banyak pula. Cara mengetahui hasil pengukuran dari alat GDL adalah dengan mencocokkan waktu yang tertera pada setiap data yang dihasilkan terhadap waktu pengukuran yang dilakukan. Pengukuran dimulai pada saat peletakkan alat, sehingga waktu yang dicatat mulai saat peletakkan alat pada tiap titik lokasi dalam ruangan. Data yang telah diperoleh dihitung rata-rata dari setiap alat yang digunakan (masing-masing alat 12 data).
b. Tekanan
Pemeriksaan perbedaan tekanan dilakukan pada hari yang sama dengan pemeriksaan particle counter. Alat yang dapat mengukur perbedaan tekanan adalah magnehelic yang dipasang di atas pintu ruangan. Ketika pintu dibuka maka perbedaan tekanan adalah nol karena antar ruangan menjadi memiliki tekanan yang sama. Hal ini disebabkan udara pada ruangan yang tekanan lebih besar mengalir ke ruangan yang tekanan lebih kecil sehingga tekanan antar ruang tersebut sama. Maka dari itu, ketika melihat magnehelic maka pintu ruangan tersebut harus ditutup rapat. Ruang yang lebih bersih memiliki tekanan yang lebih besar (positif) dibandingkan ruangan yang kurang bersih. Perbedaan tekanan antar ruangan dimaksudkan untuk menghindari kontaminasi silang. Sebab arah angin ruangan tekanan positif akan mengarah keluar ruangan sehingga ruang tersebut tetap bersih.Perbedaan tekanan antar sesama kelas adalah minimal 5 Pa sedangkan perbedaan tekanan antar ruang yang berbeda kelas menurut CPOB adalah 10-15 Pa dan menurut ISO 14644-3 Aneks B5 direkomendasikan 15 Pa. Semua ruang yang dikendalikan oleh HVAC 1 adalah kelas E umum sehingga persyaratan perbedaan tekanan antar sesama kelas adalah minimal 5 Pa.
c. Aliran udara
Pengukuran aliran udara menggunakan alat Thermo-Anemometer yang diukur dibawah supply grille (SG) pada tiap ruangan. Supply Grille pada setiap ruangan terdapat pada langit-langit ruangan. Pengukuran dalam satu supply grille dilakukan pada beberapa titik sampling. Untuk ruangan koridor, pengemasan primer dan proses gelatin, pengukuran aliran udara dilakukan pada dua supply grille yang terdapat pada ruangan tersebut. Persyaratan aliran udara untuk kelas E adalah 5-20x / jam. Untuk mendapatkan jumlah aliran udara yang besar dan tekanan yang tinggi diperlukan jenis dan model fan yang khusus dalam hal ini menggunakan sistem HVAC. Jumlah aliran udara harus disesuaikan dengan :
1. Head generation. Apabila ruang tidak menghasilkan panas yang terlalu tinggi maka tidak perlu sirkulasi yang terlalu banyak.
2. Particle generation. Apabila semakin banyak aktivitas dalam ruang yang menghasilkan partikel maka perlu sirkulasi yang cepat untuk meminimalkan kontak partikel pada ruangan.
d. Ukuran partikel
ISO 14644-1 merupakan guideline yang digunakan untuk pengukuran partikel pada ruang bersih. Standar ini melingkupi klasifikasi kebersihan ruang bersih dan zona bersih. Untuk tujuan klasifikasi ruangan menggunakan ukuran partikel 0,1 µm sampai 5 µm, sedangkan pada industri farmasi hanya mengukur jumlah partikel dengan ukuran 0,5 µm dan 5 µm. Menurut CPOB, persyaratan jumlah partikel untuk partikel 0,5 µm adalah 3450.000 sedangkan untuk partikel 5 µm adalah 29.000. Pengukuran jumlah partikel menggunakan alat Particle Counter untuk mengukur cemaran partikel per m3 setiap ruang produksi. Pengukuran setiap titik lokasi diatur selama 1 menit sebanyak 5 kali pengambilan data sehingga lama pengukuran per titik lokasi adalah 5 menit. Sebelum alat memulai pengukuran partikel, disediakan waktu selama 10 detik untuk personil berpindah tempat menjauh dari alat.
Kesimpulan
Gambaran bagaimana sistem GMP diterapkan di Indonesia dapat dilihat dari beberapa artikel yang telah terpublikasi. GMP untuk industri farmasi yang memproduksi obat mengacu pada CPOB. Topik pembahasan yang terkait industri farmasi meliputi tentang sarana penunjang berupa tata udara ruangan, pengolahan limbah, bangunan seperti area penyimpanan/gudang dan lantai, analisis kapabilitas dan kinerja mesin, analisis risiko, dan Keamanan Dan Keselamatan Kerja (K3). Publikasi yang ditemukan merupakan bagian dari aspek dari CPOB yang harus dipenuhi oleh industri farmasi, antara lain: sistem mutu produk, personel, peralatan, bangunan dan fasilitas, produksi, pengawasan mutu, gudang dan distribusi, audit, pemasok, keluhan dan penarikan produk, dokumentasi, kegiatan alih daya, kualifikasi dan validasi. Pentingnya kepatuhan industri farmasi terhadap GMP untuk memproduksi produk secara konsisten, sehingga dapat mengurangi risiko proses produksi atau menurunkan angka produk defect yang nantinya akan berpengaruh terhadap pertumbuhan profit industri farmasi.
Daftar Pustaka
1. Jagric T, Liu W, Birge J, Qin L, Sun Y, Chen H, et al. Characteristics, risk management and GMP standards of pharmaceutical companies in China. Front Public Health. 2023;01–14.
2. FDA. Pharmaceutical Quality for the 21st Century A Risk-Based Approach Progress Report. USA; 2007.
3. Haleem RM, Salem MY, Fatahallah FA, Abdelfattah LE. Quality in the pharmaceutical industry – A literature review. Vol. 23, Saudi Pharmaceutical Journal. Elsevier B.V.; 2015. p. 463–9.
4. Winarni LN. Asas Itikad Baik Sebagai Upaya Perlindungan Konsumen Dalam Perjanjian Pembiayaan. Jurnal Ilmu Hukum. 2015;11(21):1–12.
5. Presiden RI. Undang-Undang Republik Indonesia Nomor 8 Tahun 1999 Tentang Perlindungan Konsumen. Jakarta. 1999.
6. BPOM. Cara Pembuatan Obat yang Baik. Jakarta; 2018.
7. Permenkes. Permenkes NOMOR 1799/MENKES/PER/XII/2010: Industri Farmasi. Jakarta; 2010.
8. EMA. Good manufacturing practice. Amsterdam; 2023.
9. FDA. Current Good Manufacturing Practice (CGMP) Regulations. United States; 2023.
10. Ekawati AY, Husni P. Analisis Overall Equipment Effectiveness (OEE) pada Proses Pengemasan Primer di Industri Farmasi. Farmaka. 2018;16(1):27–32.
11. Sari RM, Nugraha E. Model Lean Six Sigma di Industri Farmasi. Organum: Jurnal Saintifik Manajemen dan Akuntansi. 2018;1(1):1–7.
12. Saputra R, Abdunnaser. Perancangan Instalasi Tata Udara Ruang Bersih Area Penimbangan Pada Industri Farmasi Kelas E. Bina Teknika. 2018;14(1):37–46.
13. Praditasari A, Saptarini NM. Review: Parameter Dan Metode Sampling Validasi Pembersihan Di Industri Farmasi. Farmaka. 2018;16(2):166–74.
14. Rimantho D. Analisis Kapabilitas Proses Untuk Pengendalian Kualitas Air Limbah Di Industri Farmasi. Januari. 2019;11(1).
15. Subroto W. Analisis Kapabilitas Proses Untuk Pengendalian Kualitas Air Limbah Di Industri Farmasi. Jurnal Sistem Informasi Dan E-Bisnis. 2019;1(2):39–48.
16. Susilawati S, Setiawan A, Rohayani D, Rachmawati P. Kajian Tata Udara Ruang Bersih Kelas B pada Pembuatan Obat Diabetes. Quantum Teknika : Jurnal Teknik Mesin Terapan. 2020;2(1).
17. Aditya WA, Musfiroh I. Analisis Kesesuaian Kegiatan Pergudangan dan Pemetaan Proses Pergudangan pada Salah Satu Warehouse Industri Farmasi di Jakarta. Majalah Farmasetika. 2020 May 11;5(3).
18. Hasanah TU, Wulansari T, Putra T, Fauzi M. Penerapan Lean Manufacturing dengan Metode Takt Time dan FMEA untuk Mengidentifikasi Waste pada Proses Produksi Steril PT.XYZ. Jurnal Rekayasa Sistem & Industri (JRSI). 2020 Dec 31;89.
19. Haifa AI, Permatasari NF. Pengurangan Lead Time Analisa Kemasan Primer Flexy Bag Dengan Metode Single Minute Exchange Of Dies (SMED) Di Industri Farmasi X. Jurnal Inkofar. 2020;1(1):40–6.
20. Yusuf E, Made I, Kastawan W, Pratiwi NI. Karakteristik Harmonisa Pada Sistem Daya Listrik Air Handling Unit (AHU) Industri Farmasi. Jurnal Energi. 2020;10(1):1–6.
21. Syaputra MJ. Analisa Kinerja Mesin Kemas Primer, Dengan Metode Overall Equipment Effectiveness (OEE) Di Sebuah Industri Farmasi. Journal Industrial Servicess. 2020;5(2).
22. Arif M. Analisis Kapabilitas Proses Mesin Filling Untuk Pengendalian Kualitas Pada Produk Sirup Obat Batuk Di Industri Farmasi. Jurnal Teknik Industri. 2022;95–100.
23. Nurfalah A, Saptarini NM. Evaluasi Tren Analisis Data Product Quality Review (PQR) Pada Sediaan Injeksi Methylergometrine Di Industri Farmasi A. Farmaka. 2023;21(3):282–90.
24. Junita RA, Pynkyawati T. Analisis Penggunaan Material Lantai Epoxy Pada Gedung Produksi Dan Pengemasan Vaksin Bio Farma. Jurnal Arsitektur. 2023;25(2):58–64.
25. Sukmayadi AE, Hayati Y, Rahmah AN. Uji Cemaran Mikroba Udara Pada Beberapa Ruang Produksi Non Betalaktam Lafiau Sesuai Dengan Standar CPOB Tahun 2018. Jurnal Ilmiah JKA. 2023;IX(2):9–21.
26. Mei KK, Holik A. Process Capability Study of Thiamazole 100 mg Tablet in a Pharmaceutical Industry in West Java. Journal of Pharmaceutical and Sciences. 2023;6(2):655–63.
27. Al Haitsam Dzulfiqh H, Alif Fathin M, Maulana M, Salman Al Farisi M, Desniaty R, Wijaya Abdul Rozak R. Analisis Risiko Keselamatan dan Kesehatan Kerja di Bidang Industri Farmasi (Studi Kasus Pada PT Kimia Farma Tbk. Plant Banjaran). Jurnal Ilmiah Multidisiplin. 2023;1(5).
28. Yandriyani D, Sitanggang ML, Masri I. The effect of low oveerall equipment effectiveness (OEE) on working hours and production costs of supplement products. Informatika dan Sains. 2024;14(01).
29. Elvina E, Mulyeni S. Penerapan Keselamatan dan Kesehatan Kerja di Industri Farmasi PT X. Journal of Educational Innovation and Public Health. 2024;2(1):21–34.
30. Zubair M, Maqsood S, Habib T, Usman Jan QM, Nadir U, Waseem M, et al. Manufacturing productivity analysis by applying overall equipment effectiveness metric in a pharmaceutical industry. Cogent Eng. 2021;8(1).
31. Chikwendu OC, Chima AS, Edith MC. The optimization of overall equipment effectiveness factors in a pharmaceutical company. Heliyon. 2020 Apr 1;6(4).
32. Alvira D, Helianty Y, Prassetiyo H. Usulan Peningkatan Overall Equipment Effectiveness (OEE) Pada Mesintapping Manual Dengan Meminimumkan Six Big Losses. Jurnal Online Institut Teknologi Nasional Juli. 2015;
33. Mutiara Sandy P, Wathoni N. Review: Implementation of Overall Equipment Effectiveness (OEE) Based on Lean Manufacturing Tools in the Indonesian Pharmaceutical Industry. Indonesian Journal of Pharmaceutics. 2022;4(1):158–67.
34. Omega D, Andika A. Improving Overall Equipment Effectiveness Using CPM and MOST: A Case Study of an Indonesian Pharmaceutical Company. In: IOP Conference Series: Earth and Environmental Science. Institute of Physics Publishing; 2017.
cara mengutip artikel
https://jurnal.unpad.ac.id/farmasetika/rt/captureCite/56576/23531